Identifying druggable oncogenes targeted for amplification in cancer: an introduction to the ConSig-amp analysis.
Comprehensive functional analysis of the tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers. Kim JA, Tan Y, Wang X, Cao X, Veeraraghavan J, Liang Y, Edwards DP, Huang S, Pan X, Li K, Schiff R. and Wang XS#. Nature Communications. 2016, In Press.
Genomic amplifications lead to deregulations
of oncogenes to which cancer cells become often addicted in specific
tumors. Such events, however, usually affect a large number of genes in
cancer genomes which makes it difficult to identify the primary oncogene
targets of these amplifications. In our previous study, we discovered
that cancer genes possess complicated yet distinctive “gene concept
signature”, which include cancer-related signaling pathways, molecular
interactions, transcriptional motifs, protein domains, and gene
ontologies(1). Based on this observation, we developed a Concept
Signature (or ConSig) analysis that prioritizes the biological
importance of candidate genes underlying cancer via computing their
strength of association with those cancer-related signature concepts
(http://consig.cagenome.org) (1-3). In our previous study, we have
applied this analysis to reveal the primary target genes of chromosome
17q amplifications in breast cancer (4). Here we postulate that the
ConSig analysis may be used to effectively nominate dominantly acting
cancer genes from the genomic amplifications in cancer at a genome-wide
scale, which can be further translated into viable therapeutic targets
by interrogating pharmacological databases. Indeed, analyses
of known amplified oncogene targets (i.e. ERBB2, CCND1, MYC, PAK1,
NCOA3, YWHAZ)(5-10) in breast cancer suggest that ConSig analysis can
effectively point out the primary oncogenes targeted by genomic
amplifications (Figure. 1). Toward this end, we have assembled a
genome-wide analysis called “ConSig-Amp” to discover viable therapeutic
targets in cancer from multi-dimensional genomic datasets (Figure 2a).
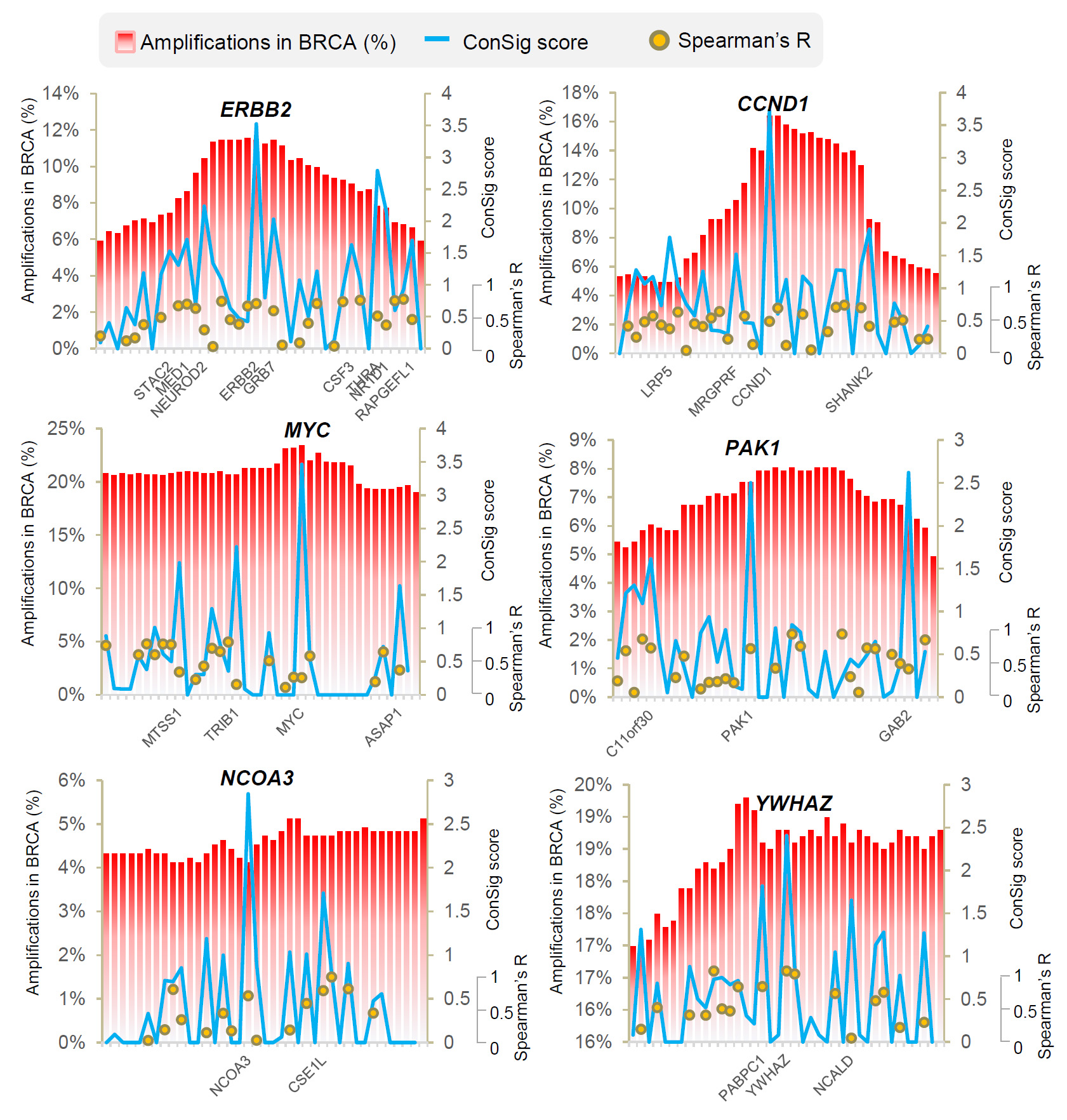
Figure 1. The
ConSig scores, the amplification frequencies, and the correlations of
expressions of the genes within the known amplified genomic regions in
breast cancer. The amplification frequencies are shown in red bar chart,
ConSig scores are shown in blue line chart, and the gene expression
correlations based on Spearman’s statistics are shown in dot-plot. The
gene names with high ConSig scores (>1.5) are shown under each chart.
To discover new therapeutic targets in ER+ breast cancer, we
analyzed the copy number (Affymetrix SNP 6.0) and RNAseq (UNC RNAseqV2)
datasets available for breast tumors from The Cancer Genome Atlas
Project (TCGA)(11). Normalized “level 3” data (segmented by the CBS
algorithm) (14) were directly applied in the analysis. First, the copy
number segments were matched with human genes based on physical
coordinates to obtain gene-level copy number data. The frequency of
genomic amplification of each human gene in breast cancer was assessed;
breast tumors with relative copy number at the respective gene locus
more than 0.7 were considered as amplification positive. Genes that are
amplified in >5% of ER+ tumors were nominated, and their expressions
based on RNAseq data were correlated with copy number data by Spearman’s
correlation statistics. The druggability of these genes was predicted
based on a drug-target database compiled from multiple sources(12-14).
Then all candidates were ranked by the ConSig-amp score calculated by
multiplying the Spearman’s correlation coefficient by the concept
signature (ConSig) score that we have developed that prioritizes
functionally important genes underlying cancer by accessing their
associations with cancer-related molecular concepts(1). The detailed
protocol to calculate the ConSig Score and the precomputed scores used
in this study (for all human genes) are available in the website
http://consig.cagenome.org (release 2).
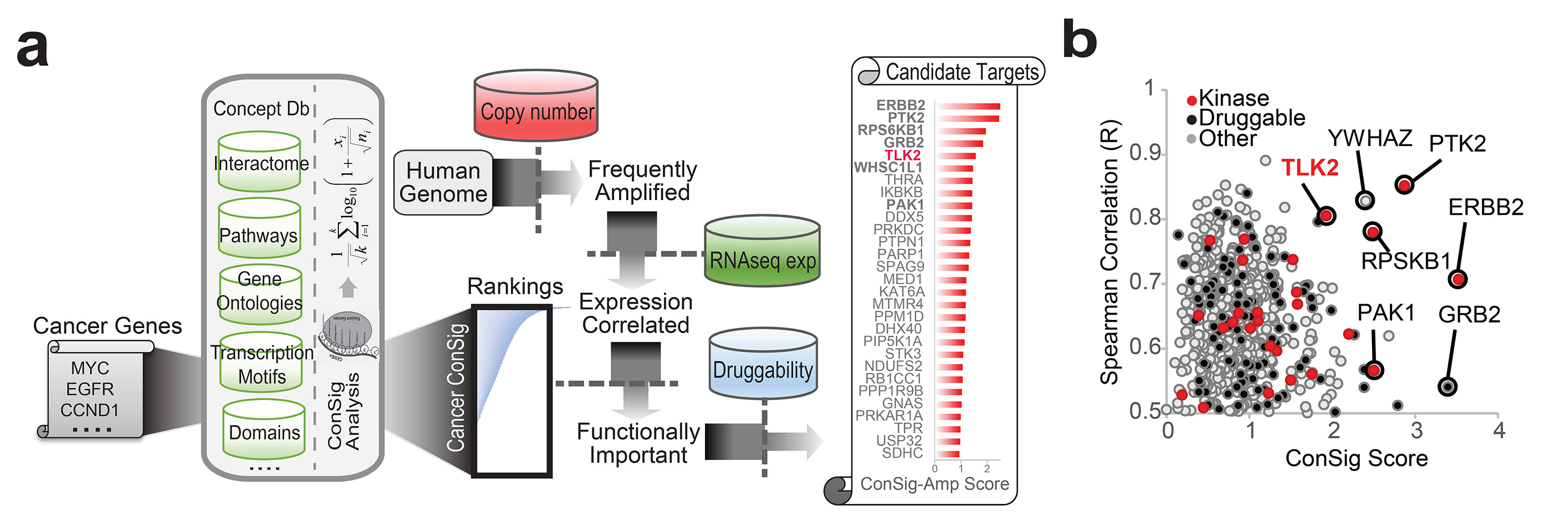
Figure 2. ConSig-Amp identifies
TLK2 as a candidate druggable target frequently amplified in breast
cancer. (a) The bioinformatics workflow of ConSig-Amp to discover
therapeutically relevant oncogene targets in cancer at genome-wide scale
based on copy number and RNAseq datasets. The ConSig-Amp score is
calculated by multiplying the ConSig score (see Methods) with the
correlation between gene expression and copy number. (b) Prioritizing
amplified breast cancer oncogene targets by ConSig score and Spearman’s
correlation between copy number (Affymetrix SNP 6.0 array) and gene
expression (RNAseq). Data shown here are from TCGA.
This analysis revealed several known kinase targets in breast
cancer such as ERBB2, PAK1, RPS6KB1, and PTK2(15, 16), together with a
new candidate kinase target, TLK2 (Figure 2b). ERBB2, RPS6KB1, and TLK2
all locate at the peaks of both frequent amplifications and high ConSig
scores in Chr17q (Figure 3). Such coincidence of the two parameters at
high levels provided integrated evidence about their role as the primary
targets of these amplified genomic regions.
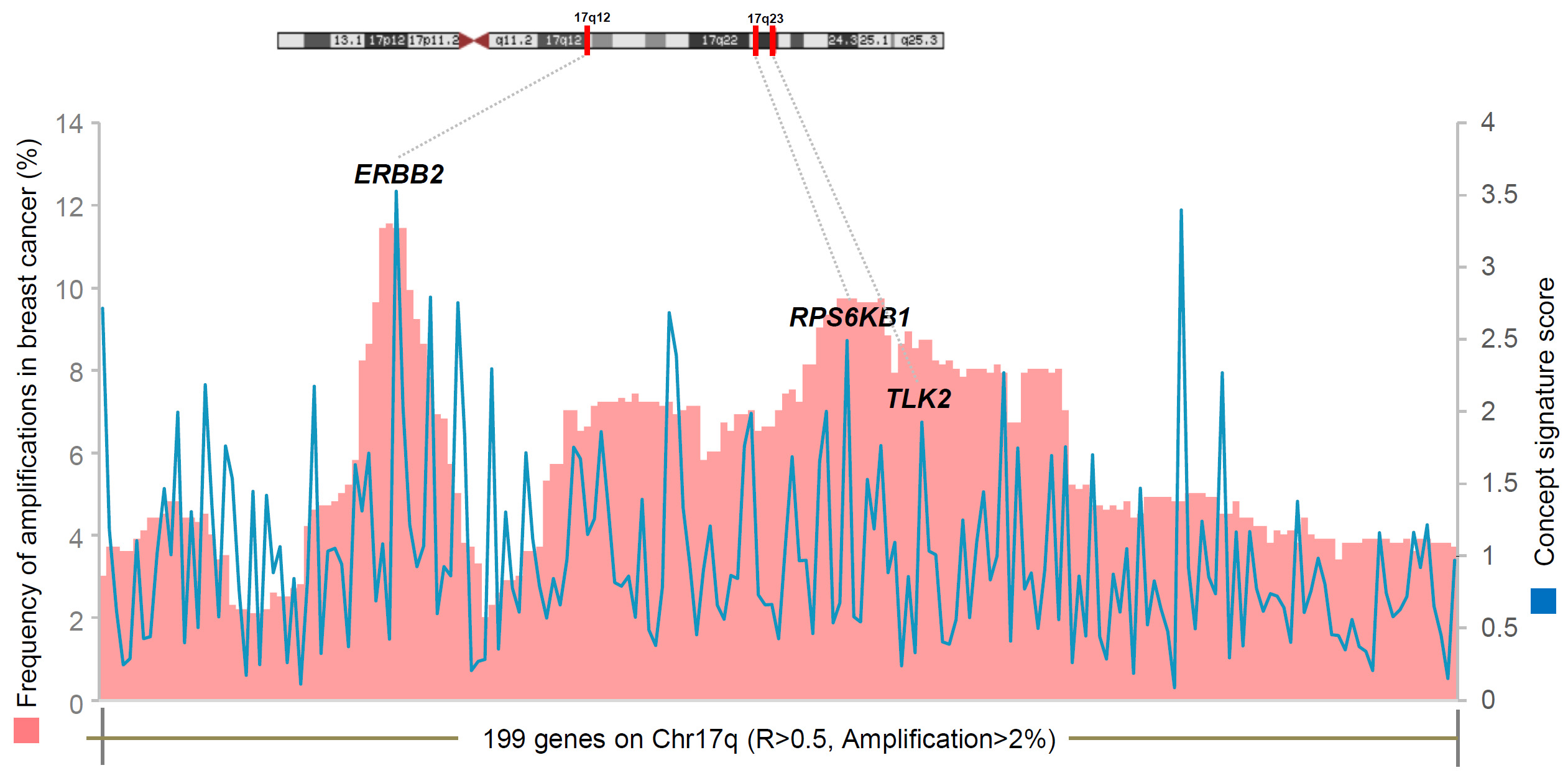
Figure
3. Frequent gene amplifications in Chr17q with significantly correlated
gene expressions. Chr17q genes amplified in >2% of breast cancers as
well as having Spearman’s correlation coefficient R>0.5 are shown in the
chart. The concept signature scores for these genes are shown in the
blue line chart. The three lead amplified kinase targets (ERBB2,
RPS6KB1, and TLK2) nominated by ConSig-Amp analysis are shown in the
chart. All three targets locate at the peaks of both genomic
amplifications and ConSig scores. This coincidence provided integrated
evidence about their functional importance in breast cancer. TLK2
locates in a small peak region of genomic amplifications close to the
RPSKB1 amplicon. This figure is based on the copy number data and RNAseq
expression data from TCGA.
Here we demonstrated the implementation of the Concept Signature
analysis, to facilitate cancer target discovery from genomic datasets.
This analysis automatically recognizes the complex molecular
fingerprints in cancer genes and enables high-throughput assessment of
the function of candidate targets underlying cancer. Interestingly,
analyses of known amplified oncogenes suggest that ConSig scores provide
independent evidence to identify oncogenes targeted by genomic
amplifications other than the correlation of copy number with gene
expression. This observation has led to our development of the
genome-wide ConSig-Amp analysis to assess the functional importance of
genes within the amplified regions of cancer genome. By interrogating
different types of genomic and pharmacological data, our integrative
ConSig-Amp analysis enables effective navigation of the complex cancer
signaling network to reveal key oncogene targets that are directly
druggable. In addition, this analysis can also be integrated with other
genomic alternations revealed by genomic or transcriptomic sequencing,
such as somatic point mutations or recurrent gene fusions, to nominate
cancer genes that are targeted by multiple types of genetic
alternations.
Applying ConSig-amp to the genomic data from TCGA has led to
our discovery of a novel cell cycle kinase target TLK2 that are
upregulated by genomic amplifications in more aggressive and lethal form
of ER+ breast cancers. This discovery suggested the application of
ConSig-Amp in discovering previously uncharacterized cancer genes
targeted for amplifications in the tumor genomes. The criteria to
determine a true gene amplification has been suggested previously:
physical mapping of the amplifications in multiple tumors, correlation
of gene expression with copy number increase, association with clinical
outcome, and its biological function in cancer(15). TLK2 gene
amplification fulfills all these criteria: a) TLK2 locates within a
consensus region of chr17q23.2 amplifications in breast cancers; b) TLK2
overexpression in breast cancer is primarily driven by increased copy
number; c) TLK2 overexpression correlates with poor clinical outcome of
ER+ breast cancer patients irrespective of endocrine treatment; d) TLK2
inhibition potently and selectively inhibits the growth of the breast
cancer cells with TLK2 amplification and overexpression; e) Our
biological studies strongly support the role of TLK2 in cell cycle
regulation, anti-apoptosis, and enhanced aggressiveness of ER+ breast
cancers.
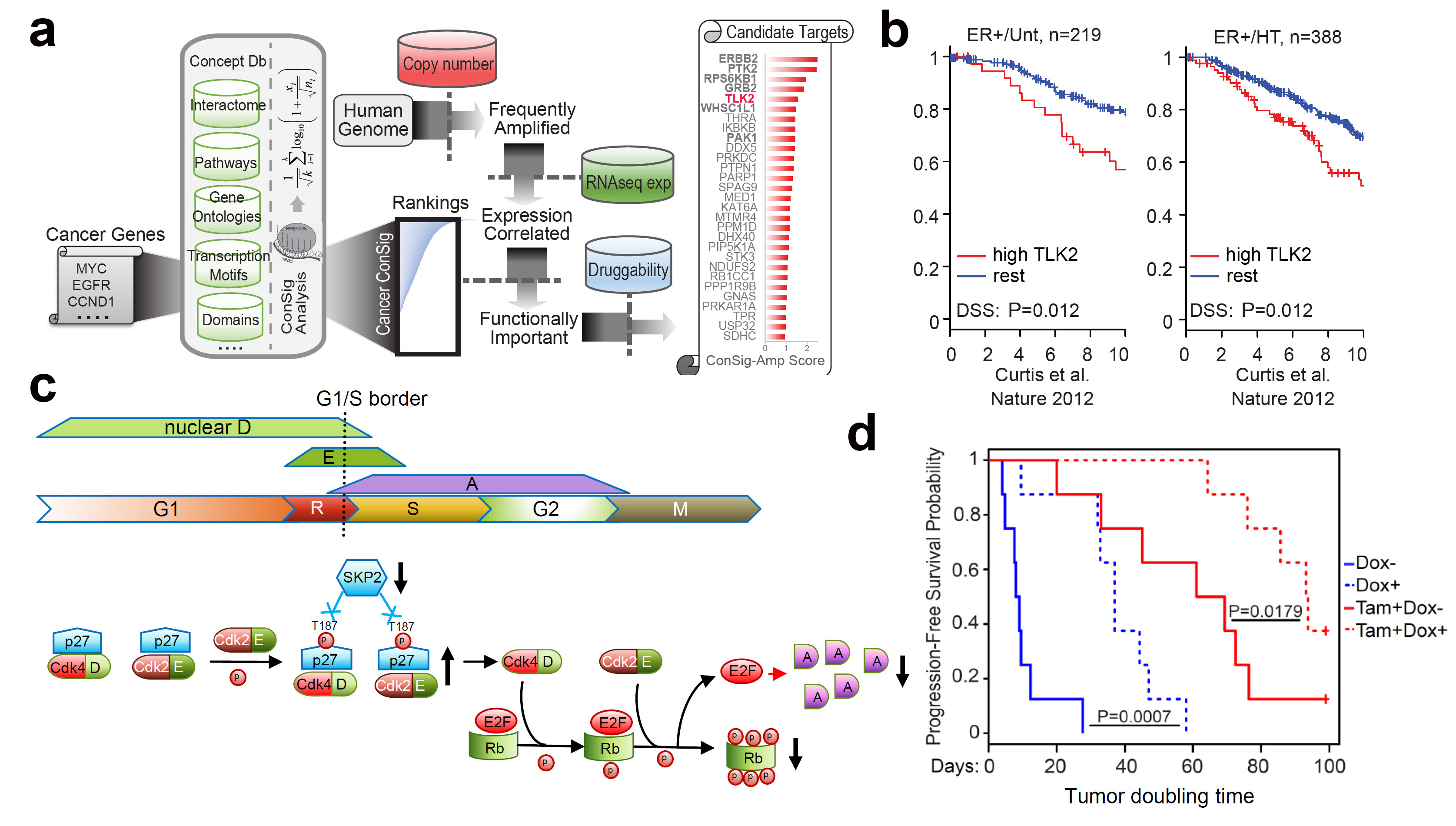
Figure 4.
Identification of TLK2 as an amplified kinase target in aggressive
luminal breast cancer. (a) The bioinformatics workflow of ConSig-Amp to
discover therapeutically relevant oncogene targets in cancer at
genome-wide scale based on TCGA copy number and RNAseq datasets. (b)
Kaplan-Meier plots based on multiple gene expression datasets showing
correlation of TLK2 overexpression with the outcome of systemically
untreated or endocrine-treated ER+ breast cancer patients. (c) A
schematic of normal G1/S cell cycle signaling and their alternations
following TLK2 inhibition (black arrows). (d) The effect of TLK2
inhibition in the MCF7 xenograft tumors inducibly expressing a TLK2
shRNA, in the presence or absence of concomitant tamoxifen treatment.
Figure shows the Kaplan–Meier survival plot comparing the
progression-free survival of different treatment groups.
Consistent with our findings, the latest
phosphoproteomic study of TCGA breast tumors by The Clinical Proteomic
Tumor Analysis Consortium (CPTAC) independently identified TLK2 as an
amplicon-associated highly phosphorylated kinases in luminal breast
cancer(17), which further support the significance of TLK2 amplification
and its preferential association with luminal tumors. Our study is the
first comprehensive analysis of TLK2 function in aggressive luminal
breast cancers, which will timely complement the CPTAC paper.
References:
1. Wang XS, Prensner JR, Chen G, Cao Q, Han B, Dhanasekaran SM, et al.
An integrative approach to reveal driver gene fusions from paired-end
sequencing data in cancer. Nat Biotechnol. 2009;27:1005-11.
2. Wang X-S, Shankar S, Dhanasekaran SM, Ateeq B, Prensner JR, Yocum AK,
et al. Characterization of KRAS Rearrangements in Metastatic Prostate
Cancer. Cancer Discovery. 2011;doi: 10.1158/2159-8274.CD-10-0022.
3. Veeraraghavan J, Tan Y, Cao XX, Kim JA, Wang X, Chamness GC, et al.
Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of
oestrogen receptor-positive breast cancers. Nat Commun. 2014;5:4577.
4. Fan Y, Ge N, Wang X, Sun W, Mao R, Bu W, et al. Amplification and
over-expression of MAP3K3 gene in human breast cancer promotes formation
and survival of breast cancer cells. J Pathol. 2014;232:75-86.
5. Borg A, Baldetorp B, Ferno M, Killander D, Olsson H, Sigurdsson H.
ERBB2 amplification in breast cancer with a high rate of proliferation.
Oncogene. 1991;6:137-43.
6. Lundgren K, Brown M, Pineda S, Cuzick J, Salter J, Zabaglo L, et al.
Effects of cyclin D1 gene amplification and protein expression on time
to recurrence in postmenopausal breast cancer patients treated with
anastrozole or tamoxifen: a TransATAC study. Breast Cancer Res.
2012;14:R57.
7. Bonilla M, Ramirez M, Lopez-Cueto J, Gariglio P. In vivo
amplification and rearrangement of c-myc oncogene in human breast
tumors. J Natl Cancer Inst. 1988;80:665-71.
8. Bostner J, Ahnstrom Waltersson M, Fornander T, Skoog L, Nordenskjold
B, Stal O. Amplification of CCND1 and PAK1 as predictors of recurrence
and tamoxifen resistance in postmenopausal breast cancer. Oncogene.
2007;26:6997-7005.
9. Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA,
et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and
HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst.
2003;95:353-61.
10. Li Y, Zou L, Li Q, Haibe-Kains B, Tian R, Desmedt C, et al.
Amplification of LAPTM4B and YWHAZ contributes to chemotherapy
resistance and recurrence of breast cancer. Nat Med. 2010;16:214-8.
11. Comprehensive molecular portraits of human breast tumours. Nature.
2012;490:61-70.
12. Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P,
et al. DrugBank: a comprehensive resource for in silico drug discovery
and exploration. Nucleic Acids Res. 2006;34:D668-72.
13. Chen X, Ji ZL, Chen YZ. TTD: Therapeutic Target Database. Nucleic
Acids Res. 2002;30:412-5.
14. Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR.
Comprehensive assay of kinase catalytic activity reveals features of
kinase inhibitor selectivity. Nature biotechnology. 2011;29:1039-45.
15. Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS. A census
of amplified and overexpressed human cancer genes. Nature reviews
Cancer. 2010;10:59-64.
16. Glenisson M, Vacher S, Callens C, Susini A, Cizeron-Clairac G, Le
Scodan R, et al. Identification of new candidate therapeutic target
genes in triple-negative breast cancer. Genes Cancer. 2012;3:63-70.
17. Philipp Mertins, D. R. Mani, Kelly V. Ruggles, Michael A. Gillette,
Karl R. Clauser, Pei Wang, et al. Proteogenomics connects somatic
mutations to signalling in breast cancer. Nature.
2016;doi:10.1038/nature18003.