|
 The principle of the Concept Signature Analysis
The principle of the Concept Signature Analysis
To
examine the key functional characteristics that assist in
new cancer gene discovery, we performed a
multi-dimensional characterization of
cancer-related gene fusions and point mutations. Placing the array
of cancer genes in the context of a compilation
of “molecular concepts”, including molecular
interactions, gene annotations and pathways
revealed the “signature concepts” defining the
genes driving cancer initiation and progression
(Figure 1).
This prompted us to generalize this finding to
develop a method that could filter non-specific
genetic aberrations in cancer. We hypothesize
that the “signature molecular concepts”
frequently found in cancer genes may be used to
define biologically meaningful genes underlying
cancer, similar to signature genes defining
certain phenotypes.
Using such information, we developed an
innovative concept signature (ConSig) technology
that nominates biologically important genetic
aberrations from high-throughput data by
assessing their association with molecular
concepts characteristic of cancer genes.
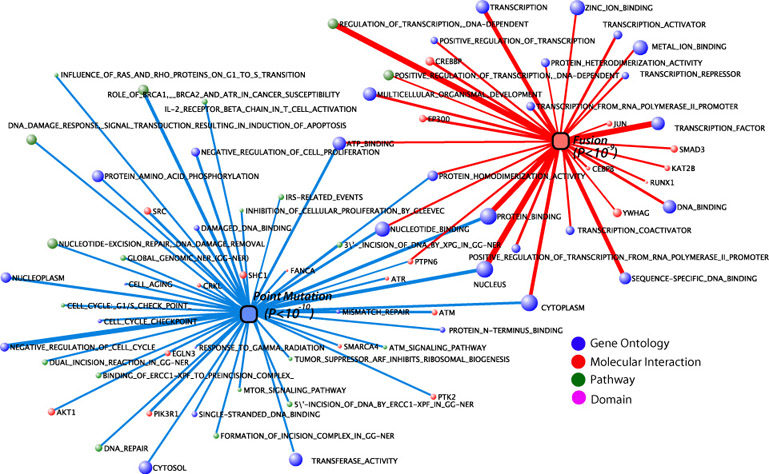
Figure 1.
Signature molecular concepts for fusion and
point mutation genes in cancer.

A
step-by-step manual for Concept Signature
Analysis
 Compile molecular concept database.
Compile molecular concept database.
In this study, we
compiled 28,963 molecular concepts from the Gene
Ontology database, the Reactome database, the
Kyoto Encyclopedia of Genes and Genomes (KEGG),
Biocarta, the HPRD database, and the Entrez Gene
conserved domain database (Table 1). To remove
redundancy, in the processing of gene
ontologies, the genes that appeared in the child
ontologies were subtracted from the parents to
avoid duplicate representation.
The compendia of non-redundant molecular
concepts used in this study can be downloaded
from
this link.
Table 1. The compendia of molecular concepts
were compiled from 6 public databases
 Compile
cancer-causal gene database.
Compile
cancer-causal gene database.
Here we use the Fusion gene list in the Mitelman
database (2008) provided by Dr. Mitelman and
mutation gene list
extracted from the
Cancer Gene Census
(2008)
as an example.
The compiled cancer causal gene lists can be
downloaded here (in NCBI Entrez Gene ID) -
[Fusion
gene list;
Point mutation gene list]
 Mapping the fusion or point mutation gene lists
against the compendia of molecular concepts.
Click the following link for the per script of
this step: [Sample
perl script 1].
Mapping the fusion or point mutation gene lists
against the compendia of molecular concepts.
Click the following link for the per script of
this step: [Sample
perl script 1].
 Calculate
the fusion and mutation ConSig-score for all
known human genes.
Calculate
the fusion and mutation ConSig-score for all
known human genes.
Computationally, let k
be the number of concepts associated with a
specified gene. Let ni
represent the number of total genes and xi
represent the number of fusion or mutation genes
participating in a given concept i, i=1,…,k.
The ConSig-score then integrates a signal
measure of fusion or mutation genes
participating in concept i (xi/ni0.5)
over all possible i, with the
incorporation of normalization factor for k
using the formula in Figure 2.
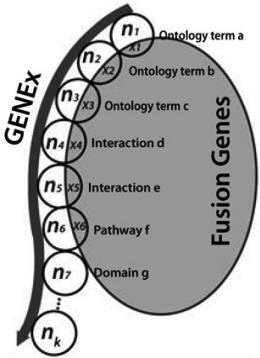 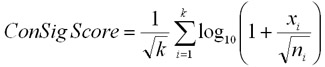 |
|
Figure 2. The algorithm for the
concept signature analysis |
With
this computation, if a gene has high probability
to be involved in gene fusions or mutations, the
fusion/mutation ConSig-score will be high
respectively; thus the radius in the
two-dimensional ConSig-score plot for fusions
and mutations will correlate with the role of
tested genes in cancer. To eliminate the bias
from the gene itself in the overlap, the seeding
genes were subtracted from the signature
concepts during the calculation of their own
ConSig score.
Download link: the perl script for this step [Sample
perl script 2].
 Calculate d- and r- ConSig score for all human
genes.
Calculate d- and r- ConSig score for all human
genes.
Plotting the fusion vs. mutation ConSig-scores
produced a striking segregation of known fusion
genes from mutation genes (Figure 3a). The
distinction line (D-line), y=1.67x, was
determined by testing optimal separation
capacity (Figure 3b), which separates 85% of mutation genes
from 80% of fusion genes. In this setting, the
radius to the zero point is defined as the
radial ConSig-score of a gene (rConSig-Score),
which indicates the strength of association with
signature concepts of both fusion and mutation
genes, thus implies the functional relevance of
candidate genes in cancer. The distance vector
from the node to the D-line, which illustrates a
distinction between fusion and mutation genes,
is defined as the distinction ConSig Score
(dConSig-Score).
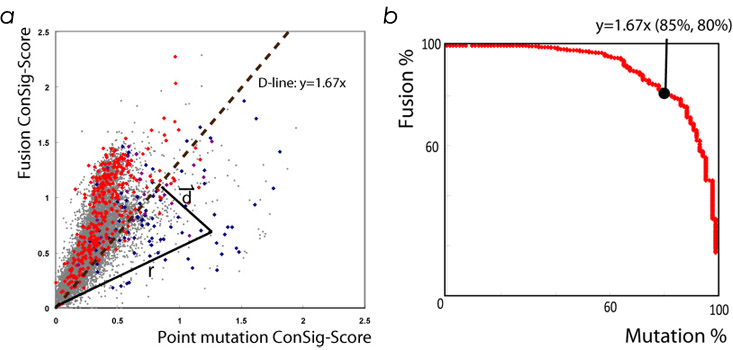
Figure 3. Plotting the fusion and mutation
ConSig score for all human genes (a) and
determining the D line (b).
Download link:
the perl script for this step
[Sample
perl script 3]
 Testing the performance of the ConSig score
ranking.
Testing the performance of the ConSig score
ranking.
Rating all human genes by the rConSig-Score will
produce substantial enrichment of established
cancer genes in top-scoring genes. Replacing the
fusion or mutation gene sets with random gene
sets produced no enrichment of the randomly
selected genes, thus validating the significance
of this observation.
The pre-computed d-
and r-ConSig score for all human genes can be
downloaded from
this link
(v2008).
|
Summary: please download the
full package
for ConSig analysis and follow the
instruction in the readme file. |
 The application of the Concept Signature
Analysis
The application of the Concept Signature
Analysis
 The discovery of driving gene fusions in cancer.
The discovery of driving gene fusions in cancer.
The ConSig technology preferentially identifies
biologically important genes in cancer. This is
particularly useful in the analysis of a large
number of putative chimeras generated by next
generation sequencing data to filter secondary
fusions. Of note, in this application, the main
theme is to evaluate the biological relevance of
putative chimeras, in stead of distinguishing
fusions and mutations, whereby the radial ConSig
will be more informative. This is especially
important for evaluating the genes involved in
both fusion and point mutations (mixed type
cancer genes), for example, a fusion involving
EGFR gene will be considered as biologically
important because of the prior knowledge of EGFR
point mutation in cancer. Moreover, the 3’
fusion partners display more distinctive
signature concepts than the 5’ partners,
therefore the ConSig technology will be more
discriminative in evaluating 3’ genes. In
practice, we usually first rate the 3’ partners
of fusion chimeras by rConSig scores, and then
rate the 5’ partners by rConSig score to
supplement this analysis.
To demonstrate the application of the above
principles, we applied the ConSig technology to
benchmark the fusion candidates detected by
paired-end transcriptome sequencing, which were
then assessed for recurrent chromosomal
aberrations using high-quality copy number data
based on the fusion breakpoint principle. While
analysis of the transcriptome data from 12 lung
cancer cell lines generated 530 putative
chimeras, the ConSig score was able to identify
the known EML4-ALK fusion as the
top-ranked candidate in the H2228 lung cancer
cell line. In addition, we found further
evidence of a R3HDM2-NFE2 fusion in H1792
cell line, which results in overexpression of
wild-type NFE2, and promotes cell
proliferation and invasion. Moreover, through
analysis of SNP arrays and lung TMAs, we find
that chromosomal rearrangements at the NFE2
locus are recurrent in a small subset of patient
tumors, suggesting that NFE2 fusion may
contribute to a new class of lung cancer biology.
 The discovery of cancer-causal point mutations.
The discovery of cancer-causal point mutations.
It is notable that in our ConSig analysis, the
point mutation genes demonstrated more
distinctive concept signatures, so that the
separation of mutation genes from the rest of
human genome is even better than fusion genes.
Therefore, the ConSig technology can be applied
to the deep sequencing data to reveal the driver
point mutations in cancer.
 The discovery of over- or under-expressed cancer
genes.
The discovery of over- or under-expressed cancer
genes.
The over- or under-expressed genes in cancer
identified from large-scale gene expression
analysis can also be ranked by ConSig score to
reveal the most biologically meaningful genes, thus nominates the most interesting
candidate for biological studies.
 The discovery of amplified or deleted cancer
genes.
The discovery of amplified or deleted cancer
genes.
The same approach also applies to the amplified
or deleted cancer genes. For example, applying
the ConSig analysis to the TCGA array CGH data for glioblastoma
revealed EGFR as top amplified oncogene candidate
in the chr 7 amplicon.
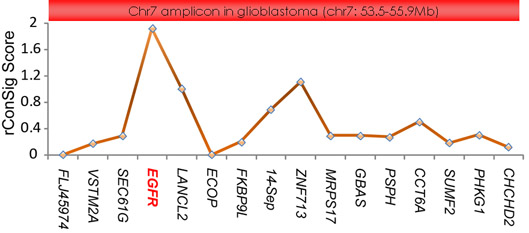
Figure 4. Revealing the primary target of Chr 7
amplicon in glioblastoma.
|