Nature Communications 2014 In Press
In this study, we develop an integrative pipeline called “Fusion Zoom” to detect recurrent gene fusions from RNAseq and genomic datasets. We postulate that the detection of pathological gene fusions would be greatly improved by applying more sensitive parameters to comprehensively capture the authentic fusion sequences from the RNAseq data, and by integrating distinct types of genomic data to prioritize the driving fusion events. Based on the observation that gene rearrangements are frequently associated with intragenic copy number aberrations (or “unbalanced” breakpoints), we previously formulated a fusion breakpoint principle to describe the characteristic intragenic copy number changes delineating recurrent fusion genes, which empowers the bioinformatics analysis to catalog meaningful fusion genes from copy number data. To facilitate high-throughput biological interpretation of candidate fusions, we also developed a Concept Signature (ConSig) analysis that nominates biologically important genes underlying cancer by assessing their association with molecular concepts characteristic of cancer genes (http://consig.cagenome.org).
Based on these principles, here we develop a pipeline that detects recurrent chimeras potentially encoding in-frame protein products from RNAseq data, catalogs the unbalanced breakpoints at the genomic loci of these fusion partner genes from copy number data, and prioritizes pathological gene fusions through the ConSig analysis. We apply this approach to the RNAseq and copy number datasets from The Cancer Genome Atlas (TCGA), and identify neoplastic fusion events between the estrogen receptor gene ESR1 and the adjacent gene CCDC170 in a subset of ER+ breast cancers that is preferentially found in luminal B tumors.
 |
Figure 1. An overview of the FusionZoom pipeline. |
Paired-end RNA sequences for 795 breast tumors and 107 paired normal breast tumors were aligned to human genome build 19 using the Tophat 2.0 fusion junction mapper. Then the putative fusion junctions were mapped to human exons (derived from UCSC gene and Ensemble gene) to identify chimerical sequences. The putative gene fusions are required to be supported by a minimum of one read that maps to the exon junctions of the two fusion genes. This criterion was expected to filter out most artifactual gene fusions randomly ligated during the sequencing procedure. This is based on the fact that authentic gene fusion junctions are usually formed by exon boundaries of partnering genes, whereas the fusion junctions of these artifactual fusions are unlikely to coincide with exon boundaries.
Putative fusion sequences were then constructed and aligned against human genome and transcriptome using the accurate aligner BLAST. The chimeric sequences that can mostly align to a wild-type genomic or transcript sequence were disregarded. After such filtering, a total of 68,611 chimeras with >2 median number of reads across all tumors were identified. A total of 2790 putative fusions were identified as somatic and recurrent (present in more than one breast tumors).
Next, we went on to analyze the potential of these fusion candidates to encode in-fram protein products. Here the in-frame analysis detects a fusion that either results in an in-frame chimerical protein, or combines the untranslated 5’ UTR of the 5’ partner with the full-length ORF of the 3’ partner. This is computed based on the reading-frames of the respective UCSC and Ensemble wild-type transcripts. The ORF analysis based on the reading frames of exons of the partner genes cannot predict all the de novo ORFs generated by the fusions. Here we required the candidate fusion to present an in-frame fusion variant in at least one sample. This step filtered out about 1000 candidates that never present any in-frame variant in any single sample, which are less likely to be functionally relevant.
To assess the unbalanced breakpoints within candidate fusion genes, we obtained TCGA “level 3” Affymetrix SNP 6.0 copy number data for 865 breast tumors. These level 3 data are generated by circular binary segmentation. The genomic position of each copy number transition was mapped with the genomic regions of all human genes. The genomic region of each human gene was designated as the starting of the transcript variant most approaching the 5’ of the gene, and the end of the variant most approaching the 3’ of the gene. The “broken” genes with intragenic copy number breakpoints were classified into candidate 5’ and 3’ partners based on the association of these unbalanced breakpoints with gene placements. 5’ amplified genes or 3’ deleted genes were considered as potential 5’ partners, while 5’ deleted or 3’ amplified genes were considered as potential 3’ partners according to the fusion breakpoint principle.
The fusion candidates were then ranked by the incidence of fusion transcripts in breast tumors, the concept signature (ConSig) score (http://consig.cagenome.org, release 2), and the recurrent unbalanced breakpoints at the genomic loci of fusion genes.
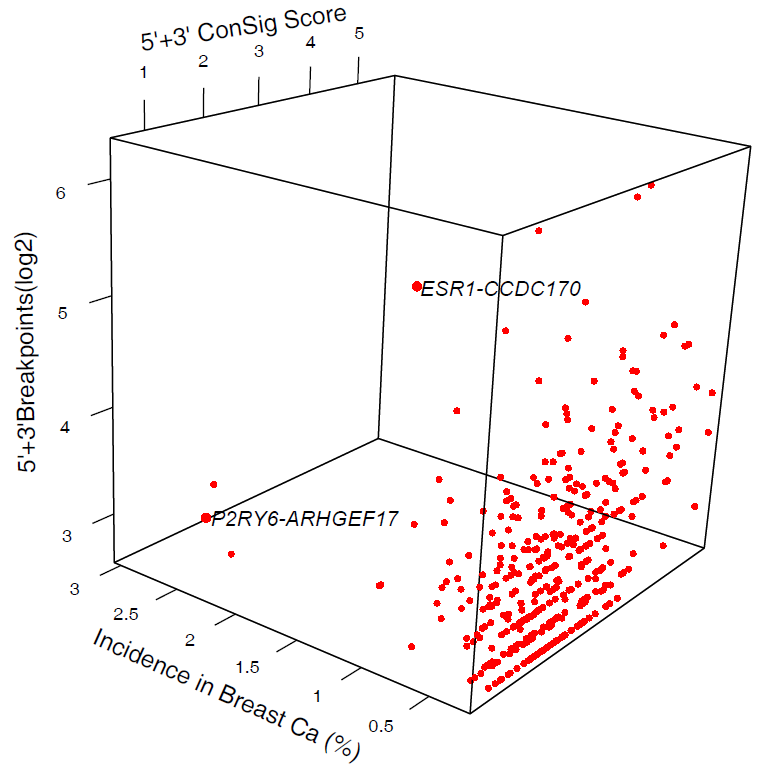 Click here to see the plot in interactive 3D mode (please enable java) |
Figure 2. Prioritizing the fusion candidates by the incidence of candidate fusions in breast tumors, the concept signature (ConSig) score, and the recurrent unbalanced breakpoints at the genomic loci of fusion genes. The data shown here correspond to Supplementary Data 1. |
Note: The copy number and RNAseq (Illumina HiSeq, paired-end) data for breast tumors used in this study were from TCGA (http://cancergenome.nih.gov/ and https://cghub.ucsc.edu).
The discovery alignments can catelog the fusion reads from RNAseq data in a genome-wide scale. However, the fusion junction reads detected by the discovery alignments are often subject to bias of misalignments. After nominating the lead recurrent fusion candidate, it will be necessary to reconstruct all possible fusion sequences for the lead fusion and align these putative sequences with the RNAseq reads. This will more accurately capture the fusion reads from RNAseq data for the lead candidate, and thus more comprehensively evaluate its incidence.
To more accurately capture ESR1-CCDC170 chimerical reads, we reconstructed all putative fusion-variant transcripts by combining each of ESR1 exons with each of the CCDC170 exons. The resulting putative ESR1-CCDC170 variant sequences are provided in Supplementary Data 2. Using the Burrows-Wheeler Aligner (BWA), we aligned these ESR1-CCDC170 variant sequences with the RNAseq data for 990 breast tumors released to date by TCGA, allowing up to 3 mismatches. Using a Perl script, we processed the Bam output files to identify junction or encompassing chimerical reads. A series of filtering steps were performed to remove the false positives due to misalignments.
To assure that the alignments are acceptable, paired reads supporting ESR1-CCDC170 were manually realigned with the respective putative variant sequences as well as the human transcriptome and genome reference sequences using BLAST or BLAT. The raw sequences of fusion reads identified after these filtering are provided in Supplementary Data 3. A breast tumor was considered as fusion-positive if BWA revealed a minimum of three chimerical reads with at least one read mapped to the fusion junction.