Research Projects
The overview
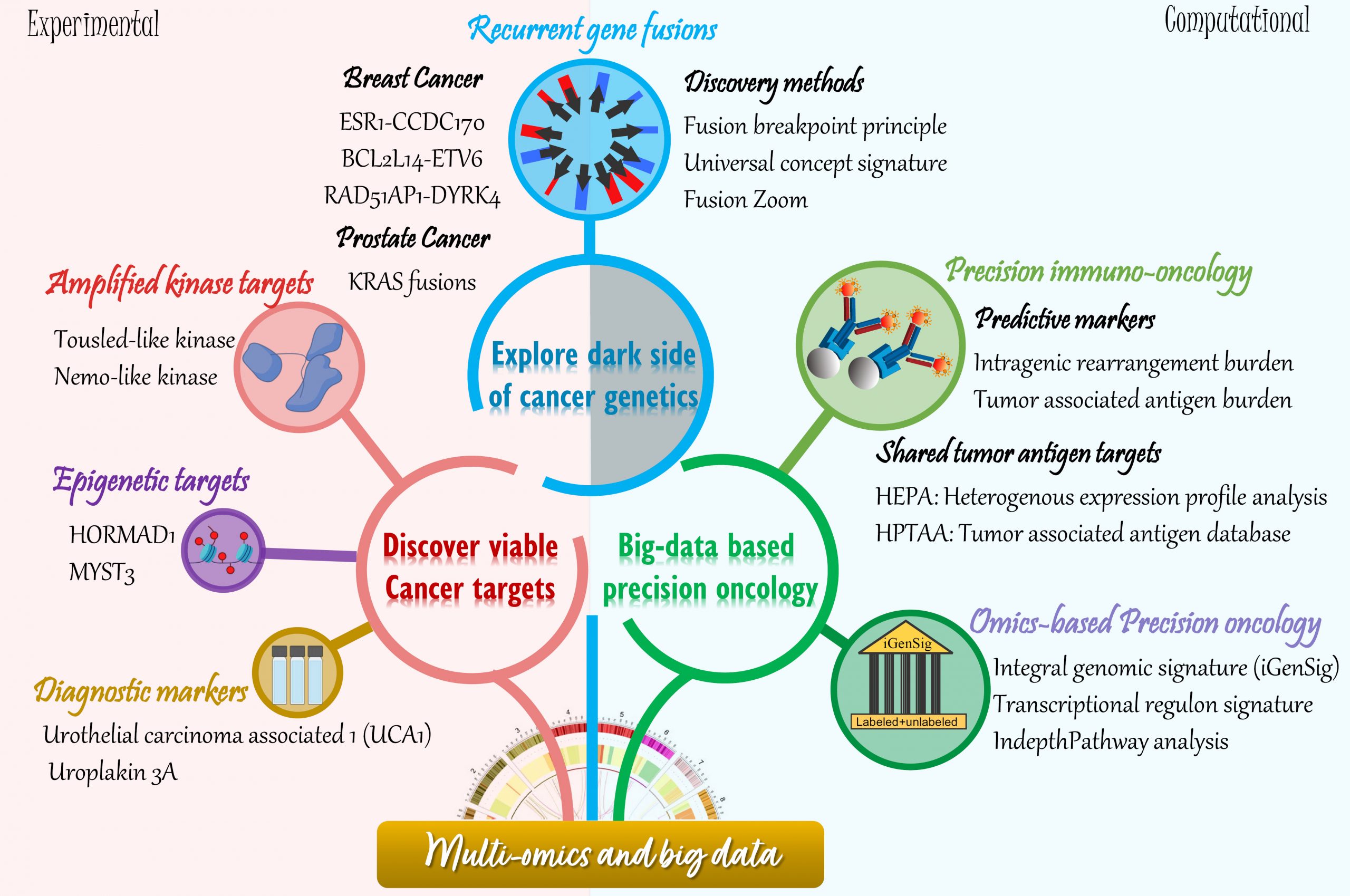
Our research projects seamlessly integrate computational and laboratory studies. Our wet laboratory research focus is to explore the “dark-side” of breast cancer genetics for identification of novel therapeutic targets and predictive biomarkers, with aims to develop precision therapeutics. By interrogating multidimensional genomic datasets, we have identified three recurrent gene fusions and two kinase targets deregulated by genomic rearrangements or amplifications in aggressive breast cancers. The clinical relevance and biological function of these targets, their role in breast cancer therapeutic resistance, as well as the potential therapeutic strategies to more effectively manage these fusion-associated carcinomas of the breast are being investigated. We expect that our new discoveries will yield novel insights into recurring genetic abnormalities leading to breast cancer, and establish robust targets for effective and personalized therapies. Our dry lab projects focus on developing computational technologies to discover driver cancer genes and targets as well as modeling therapeutic responses based on multi-OMIC datasets. In particular, our lab pioneered a new class of multi-omics modeling methods called “integral genomic signature analysis” for big data based precision oncology, and developed a computational approach called HEPA for high-throughput identification of tumor associated antigen targets.
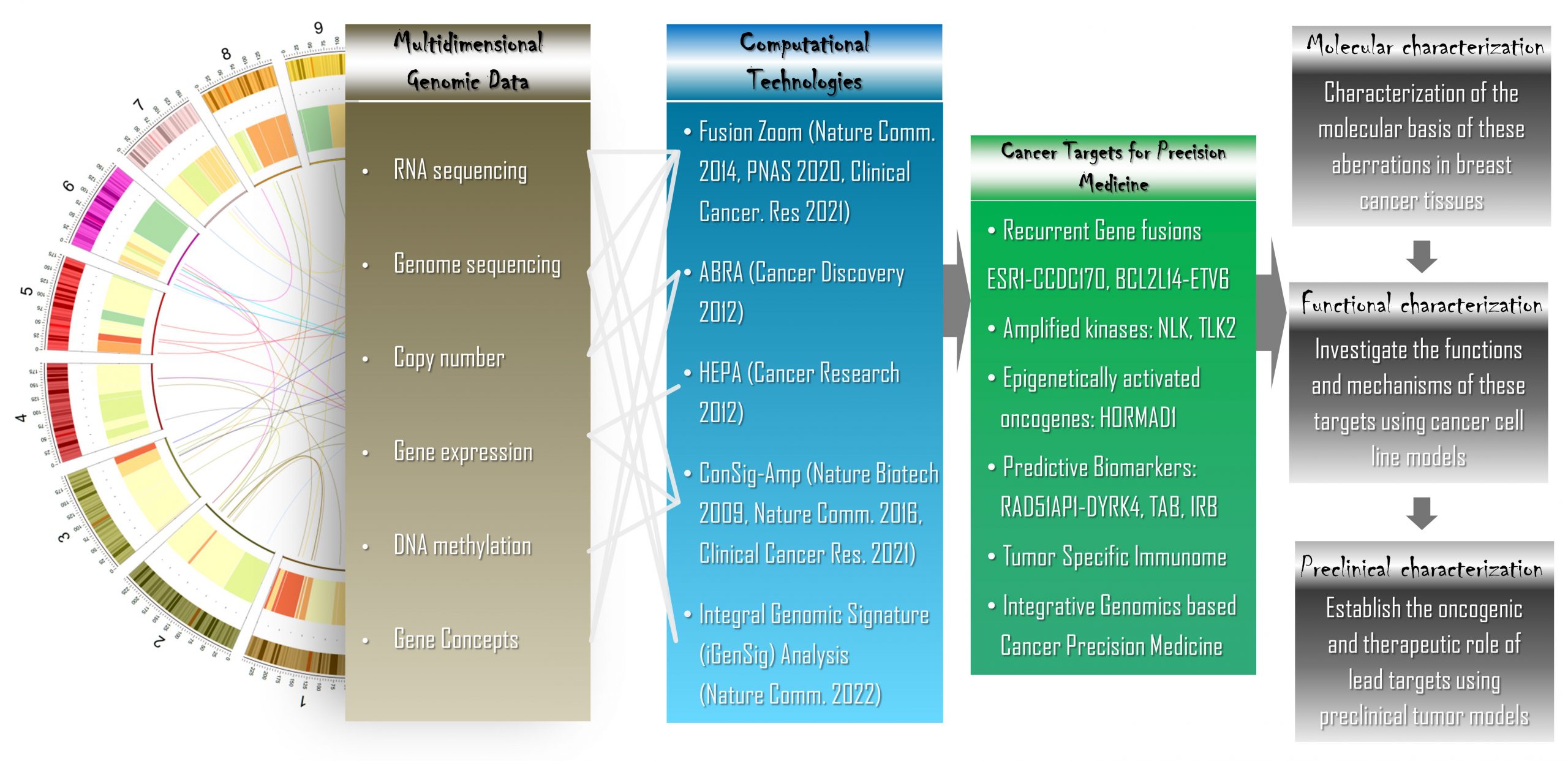
An integrated computational and laboratory approach to translate multidimensional genomic data into viable cancer targets and precision therapeutics
These projects have been funded by the National Cancer Institute (two R01 awards, and one R21 award), Department of Defense Congressionally Directed Medical Research Programs (two breakthrough Awards [Level 1 and 2], Idea Award and two Postdoctoral Fellowship Awards), Susan G. Komen for the Cure Foundation (two Postdoc Fellowship awards), PA Breast Cancer Coalition, Nancy Owens Memorial Foundation, Breast Cancer Research Foundation, the Shear Family Foundation, the Hillman Foundation, and Commonwealth of PA.



Explore the dark-side of cancer genetics
I. An integrative approach to discover pathological recurrent gene fusions in solid tumors
Recurrent gene fusions represent a critical class of oncogenic aberrations that have helped shape the precision medicine landscape. The EML4-ALK and ROS1 fusions found in 4-5% and 1-2% of lung cancer cases respectively have been targeted with effective novel therapies resulting in subsequent durable disease control. Recently, Larotrectinib has received FDA approval as the first targeted therapy with tissue-agnostic indications against Neurotrophic Tyrosine Receptor Kinase (NTRK) fusions found in ~1% of solid tumors. To examine the key characteristics that assist in the discovery of recurrent gene fusions in solid tumors, we performed a multi-dimensional characterization of known cancer-related gene fusions. Placing the array of cancer genes in the context of a compilation of “molecular concepts”, including molecular interactions, gene annotations and pathways revealed the “signature concepts” defining the genes driving cancer initiation and progression. Using such information, we developed an innovative concept signature (ConSig) technology that nominates biologically important genetic aberrations from high-throughput data by assessing their association with molecular concepts characteristic of cancer genes. To integrate use of high-throughput genomic data, we analyzed the genomic imbalances associated with known gene fusions, finding that recurrent gene fusions exhibit distinctive patterns of copy number alterations corresponding to differential portions of fusion partners, which we formulated as the “fusion breakpoint principle”. Based on these principles, we then developed a powerful integrative bioinformatics pipeline called “Fusion Zoom” to reveal recurrent pathological gene fusions in cancer from transcriptome sequencing data (Nature Biotechnology 2009. Read More.).
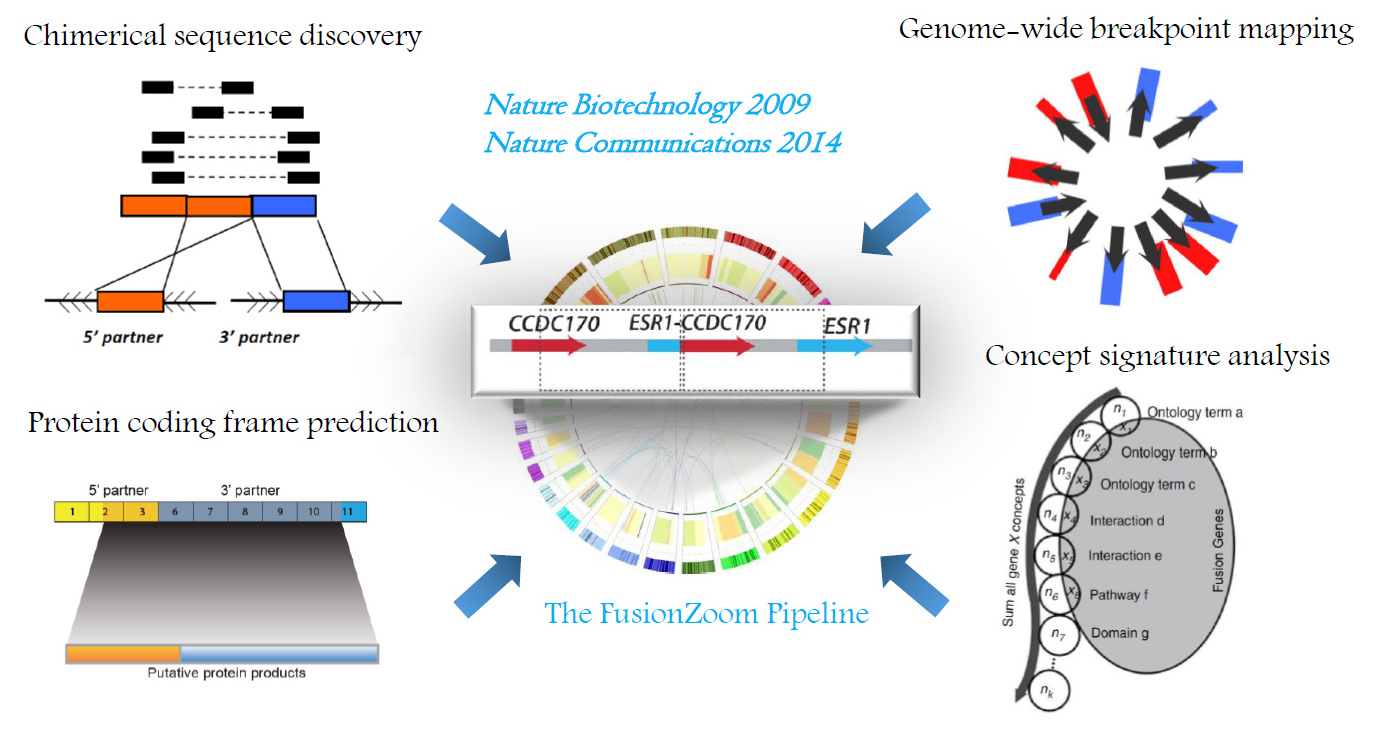
“Fusion Zoom”: revealing pathological recurrent gene fusions from the scrambled cancer genome.
Using this approach, we discovered recurrent KRAS rearrangements in metastatic prostate cancer. The most significant KRAS fusion, UBC2L3-KRAS, generates a chimeric protein, which attenuates canonical MEK/ERK signaling, activates AKT and p38 MAPK pathways, and promote cell invasion and xenograft growth. This is the first report of a gene fusion involving Ras family suggesting that this aberration may drive metastatic progression in a rare subset of prostate cancers (Cancer Discovery 2012. Read More.).
II. Discover recurrent pathological gene fusions in more aggressive and therapy-resistant breast cancers
A. Recurrent ESR1-CCDC170 fusions in more aggressive, endocrine-resistant, and metastatic luminal breast cancers
In 2014, our lab reported the first ESR1 fusions generated by localized rearrangements between the ESR1 gene encoding ERα and its immediate centromeric neighbor gene coiled-coil domain containing 170 (CCDC170)(Nature Communications. 2014. Read More.). ESR1-CCDC170 fusions are detected in ~8-11 % of luminal B, endocrine-resistant, and metastatic ER-positive breast cancers, a majority of which are tandem duplications. Most ESR1-CCDC170 fusions join the 5’ untranslated region of ESR1 to the coding region of CCDC170 resulting in the expression of an amino(N)-terminally truncated CCDC170 driven by a constitutively active ESR1 promoter. To date, ESR1-CCDC170 remains the most frequent pathological gene fusion detected in luminal breast cancer, and its recurrence has been subsequently supported by multiple clinical studies (Genes Chromosomes Cancer. 2022. Read More.). A neoadjuvant clinical trial suggested that ESR1-CCDC170 fusions were preferentially detected in endocrine resistant or intermediate resistant luminal breast tumors (Science Translational Med. 2017). This fusion is also reported as a recurrent event in ovarian cancer which only exists in patients with exceptional short-term survival. Consistent with the clinical observations, our data suggest that ESR1-CCDC170 endows increased cell motility, invasion, and anchorage-independent growth in vitro, and enhanced tumor formation in vivo.
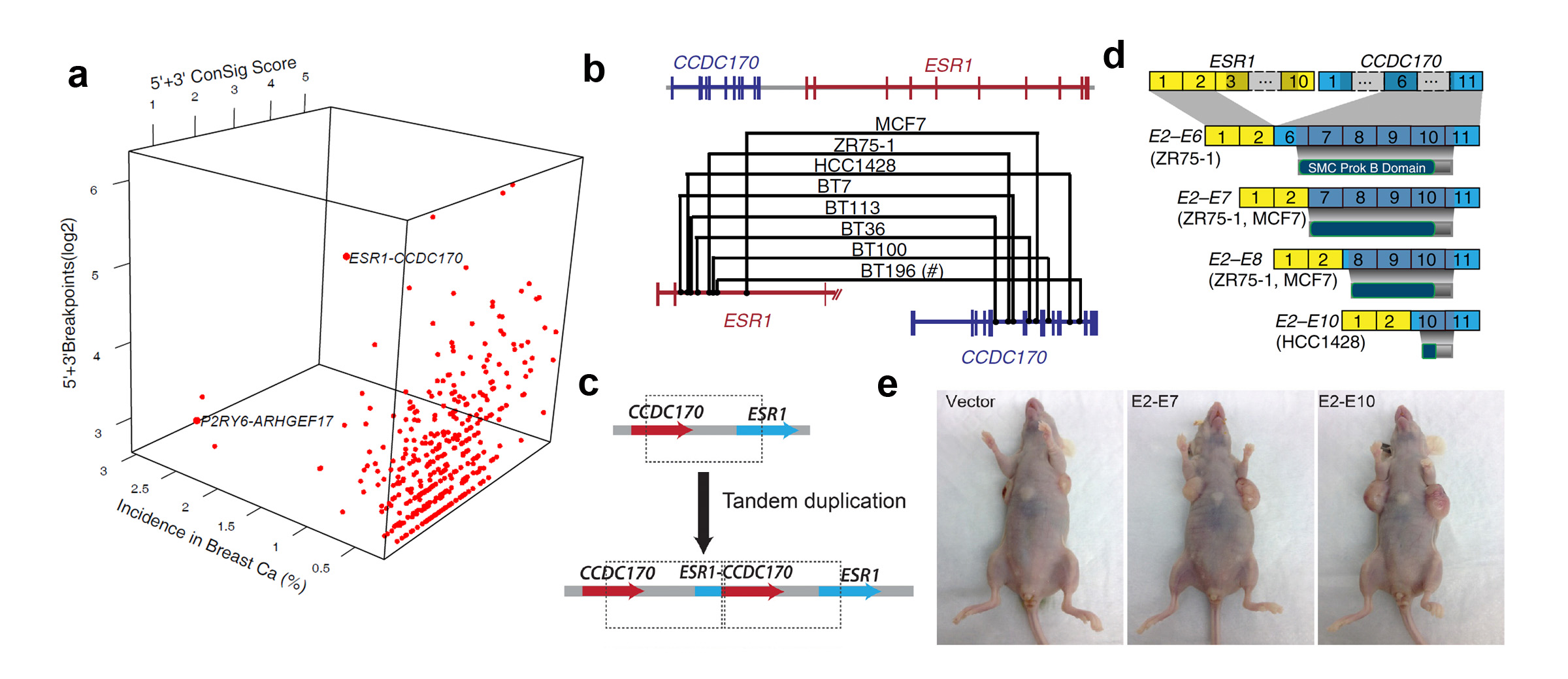
Discover and characterize recurrent ESR1-CCDC170 fusions in an aggressive subset of estrogen-receptor positive breast cancers. (a) FusionZoom identifies ESR1-CCDC170 as the lead pathological fusion candidate from RNAseq and copy number data. (b) Genomic fusion points identified by genomic PCR in ESR1-CCDC170 positive tumors. (c) ESR1-CCDC170 may be generated by tandem duplications. (d) ESR1-CCDC170 fusions produce truncated CCDC170 proteins. (e) ESR1-CCDC170 enhanced tumor formation in nude mice when introduced into the breast cancer cells.
Our most recent study suggests that ESR1-CCDC170 fusions may endow breast cancer cell survival under endocrine stress via physical binding of the fusion to the HER2/HER3/SRC complex and activation of downstream signaling pathways (Breast Cancer Research 2021. Read More.). This suggest that ESR1-CCDC170 may engender a unique exploitable vulnerability in these lethal tumors that can be potentially targeted by HER2/SRC inhibitors. Supported by our recent award from PA Breast Cancer Coalition, we are investigating the potential therapeutic strategy to better manage endocrine resistant and metastatic breast cancers harboring this fusion.
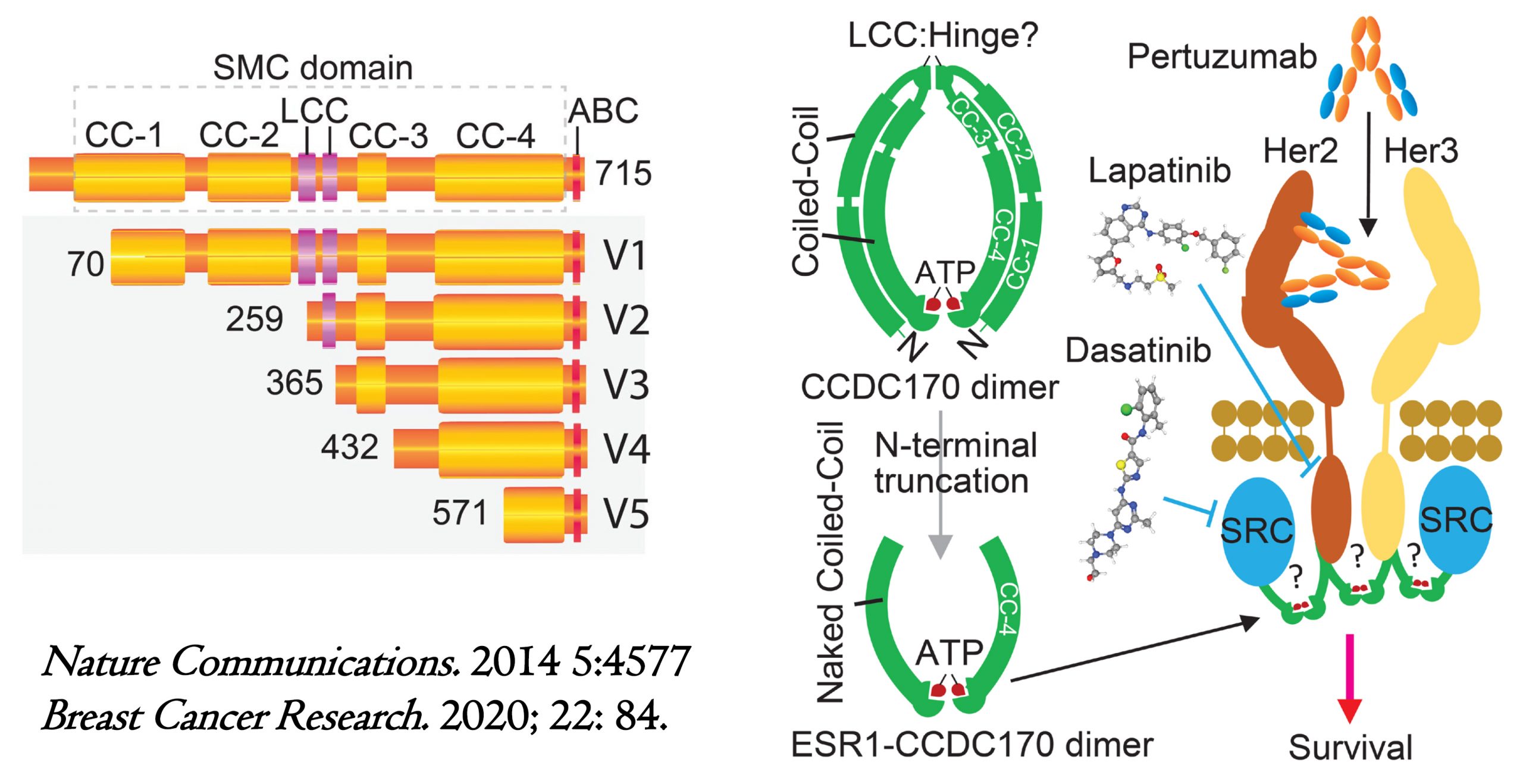
Protein variants and action mechanisms of ESR1-CCDC170 fusions in luminal breast cancer. (A) Schematic of ESR1-CCDC170 fusion proteins. CC, Coiled-coil; ABC: ATP-binding cassette, LCC, Low compositional complexity. (B) Schematic showing the hypothesized mechanism engaged by ESR1-CCDC170 to endow breast cancer cell survival and the druggable hypothesis.
B. Recurrent BCL2L14-ETV6 fusions in more aggressive and chemo-resistant triple-negative breast cancer
Our recent landscape study of triple-negative breast cancer genetics revealed a recurrent gene fusion involving the prototype cancer gene ETV6 and its immediate telomeric neighbor gene BCL2L14, which is exclusively detected in 6-12% of TNBC according to WGS data. Clinically BCL2L14-ETV6 associated TNBC breast tumors tend to show necrosis areas, a higher tubule formation score, and higher nuclear pleomorphism. Amid TNBC subtypes, BCL2L14-ETV6 is positive in ~19% of these tumors and most frequently detected in the mesenchymal TNBC subtype. Subsequent experimental studies suggest that BCL2L14-ETV6 fusions enhance cell motility and invasiveness, prime EMT, and endow paclitaxel resistance of TNBC75. While the fusion variants encode diverse open-reading frames that encode either chimerical proteins or truncated BCL2L14 proteins, these variants appear to induce similar gene expression and phenotypic changes distinctive from wildtype ETV6. This is the first report of a TNBC-specific recurrent gene fusion and the first report of a 3’ ETV6 gene fusion in solid tumors. Supported by our recent level 2 breakthrough award from the Department of Defense, we are studying the function of BCL2L14-ETV6 in immune evasion and the therapeutic implications. Future studies of BCL2L14-ETV6 induced EMT, taxane resistance, and immune evasion will help establish its role as a genomic determinant of taxane sensitivity and reveal optimized chemotherapy regimens as well as new therapeutic vulnerability of BCL2L14-ETV6 positive tumors (PNAS 2020. Read More.)
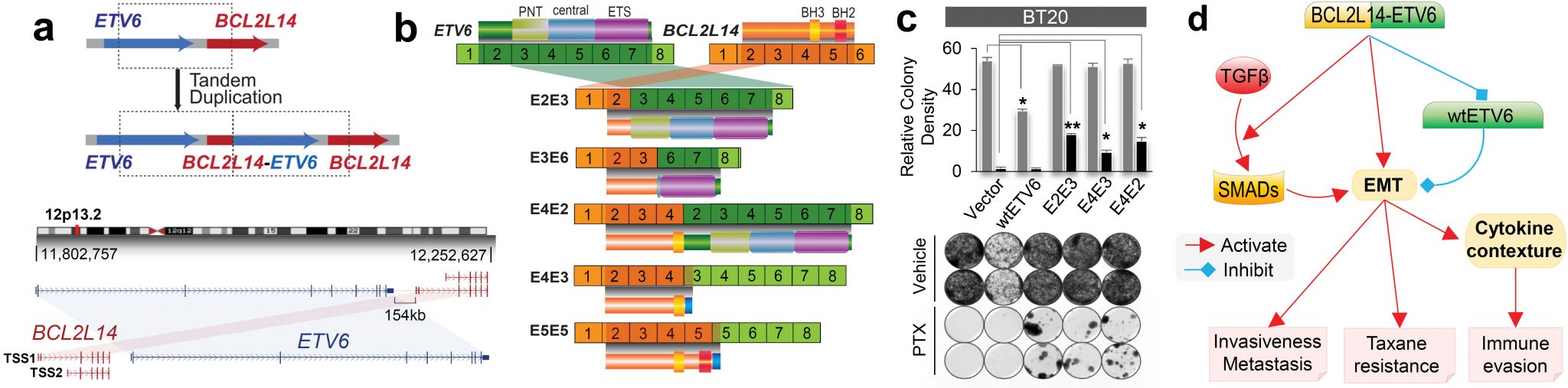
BCL2L14-ETV6 fusion drives epithelial-mesenchymal transition and endows more aggressive and therapy-resistant triple-negative breast cancer. (a) Schematic of the tandem duplication generating the BCL2L14-ETV6 fusions (upper panel) and schematic of BCL2L14/ETV6 genomic loci. TSS: transcription start sites (lower panel). (b) Schematic of BCL2L14-ETV6 and ETV6 IGR variants and encoded proteins. (c) BCL2L14-ETV6 fusions endow clonal resistance in BT20 cells following paclitaxel treatment as shown by clonogenic assay. (d) Hypothesis about the action mechanisms of BCL2L14-ETV6 fusion to drive more aggressive and therapy-resistant TNBC.
C. RAD51AP1-DYRK4 fusions confer sensitivity to MEK inhibitors in more aggressive luminal breast cancers
In addition to genetic fusions, our most recent large-scale analysis of TCGA RNAseq data discovered a neoplastic fusion transcript (RAD51AP1-DYRK4) preferentially overexpressed in luminal B breast cancers (7-17.5%). This fusion is a nontraditional fusion transcript generated by cancer-testis specific intergenic splicing. RAD51AP1-DYRK4 encodes a C-terminally truncated RAD51AP1 protein fused to a frame-shift peptide derived from DYRK4 and resulting in a cytoplasmic-localized chimeric protein that retains the intrinsic disordered (ID) regions but lacks the RAD51-interacting domain. RAD51AP1-DYRK4 appears to rewire key oncogenic signaling and induce cancer cell addiction to MEK/ERK signaling through attenuating PI3K/AKT cascades and thus could constitute as an Achilles heel to attack these deadly breast cancers by conferring increased sensitivity to the MEK inhibitor trametinib. The discovery of RAD51AP1-DYRK reveals a new class of pathological molecular events accountable for the increased aggressiveness and metastasis-prone behavior of lethal breast cancer forms (Clinical Cancer Research 2021. Read More.).
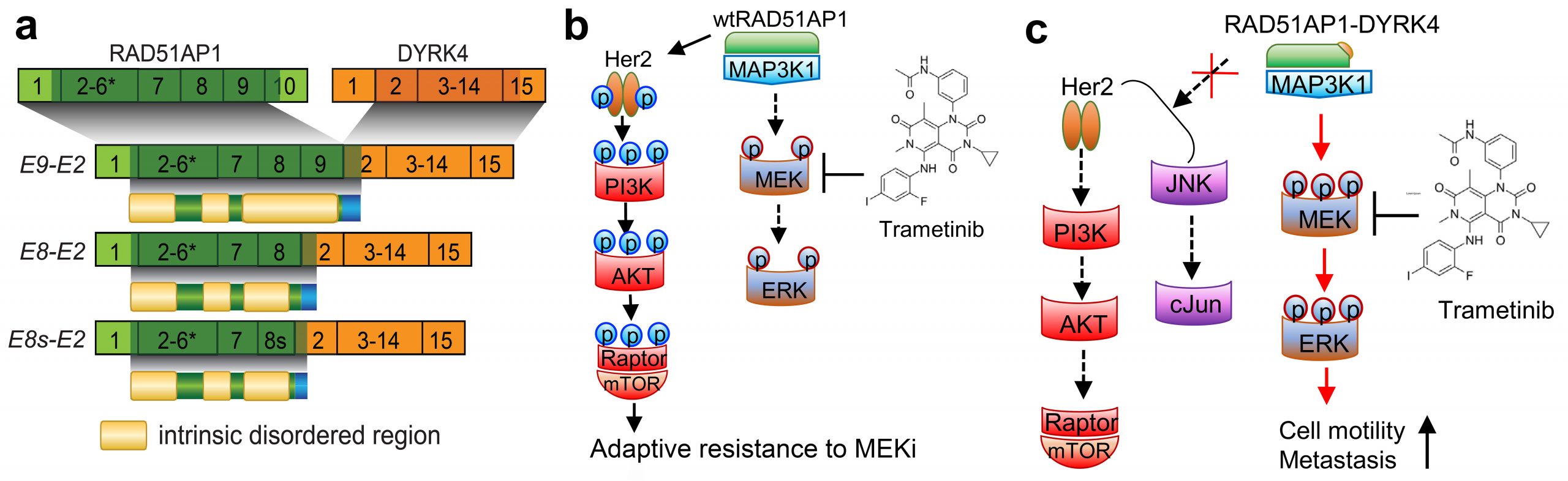
RAD51AP1-DYRK4 fusions confer MEK inhibitor sensitivity via attenuating the compensatory feedback loop following MEK inhibition. (a) Schematic of RAD51AP1-DYRK4 major fusion variants and their encoded proteins identified in breast cancer cell lines and tissues. (b-c) The mechanism engaged by RAD51AP1-DYRK4 to endow increased aggressiveness and confer sensitivity to MEK inhibition. RAD51AP1-DYRK4 forms complex with MAP3K1, activates MEK/ERK, and attenuates HER2/PI3K/AKT and JNK/c-Jun cascades under MEK inhibition (c). In contrast, wtRAD51AP1 overexpressing cancer cells show compensatory activation of the HER2/PI3K/AKT under MEK inhibition, leading to adaptive resistance to trametinib (b).
Discover viable cancer targets for the development of precision therapeutics
A. Discovery of tousled-like kinase 2 frequently amplified in aggressive luminal breast cancers
More aggressive and therapy-resistant ER-positive breast cancers remain a great clinical challenge. To identify new kinase targets for effective intervention, we applied our integrative ConSig-amp analysis to the multi-dimensional genomic datasets from The Cancer Genome Atlas. This analysis revealed tousled-like kinase 2 (TLK2) as a lead candidate kinase target that is frequently amplified in ~10.5% of ER-positive breast tumors. The resulting overexpression of TLK2 is more significant in aggressive and advanced tumors, and correlates with worse clinical outcome regardless of endocrine therapy (Figure 4b). Ectopic expression of TLK2 leads to enhanced aggressiveness in breast cancer cells, which may involve the EGFR/SRC/FAK signaling. Conversely, TLK2 inhibition selectively inhibits the growth of TLK2-high breast cancer cells, downregulates ERα, BCL2, and SKP2, impairs G1/S cell-cycle progression (Figure 4c), induces apoptosis, and significantly improves progression-free survival in vivo (Figure 4d). We have identified two potential TLK2 inhibitors that could serve as backbones for future drug development. This study represents the first comprehensive analysis of TLK2 function in aggressive luminal breast cancers (Nature Communications. 2016. Read More.).
In addition, we discovered that TLK2 overexpression mechanistically impairs Chk1/2-induced DNA-damage checkpoint signaling, leading to a G2/M checkpoint defect, delayed DNA repair process, and increased chromosomal instability. This is the first observation linking TLK2 function to chromosomal instability. This finding yields new insight into the deregulated DNA damage pathway and increased genomic instability in aggressive luminal breast cancers (Mol. Cancer Res. 2016. Read More.). Together, amplification of TLK2 presents an attractive genomic target for aggressive ER-positive breast cancers.
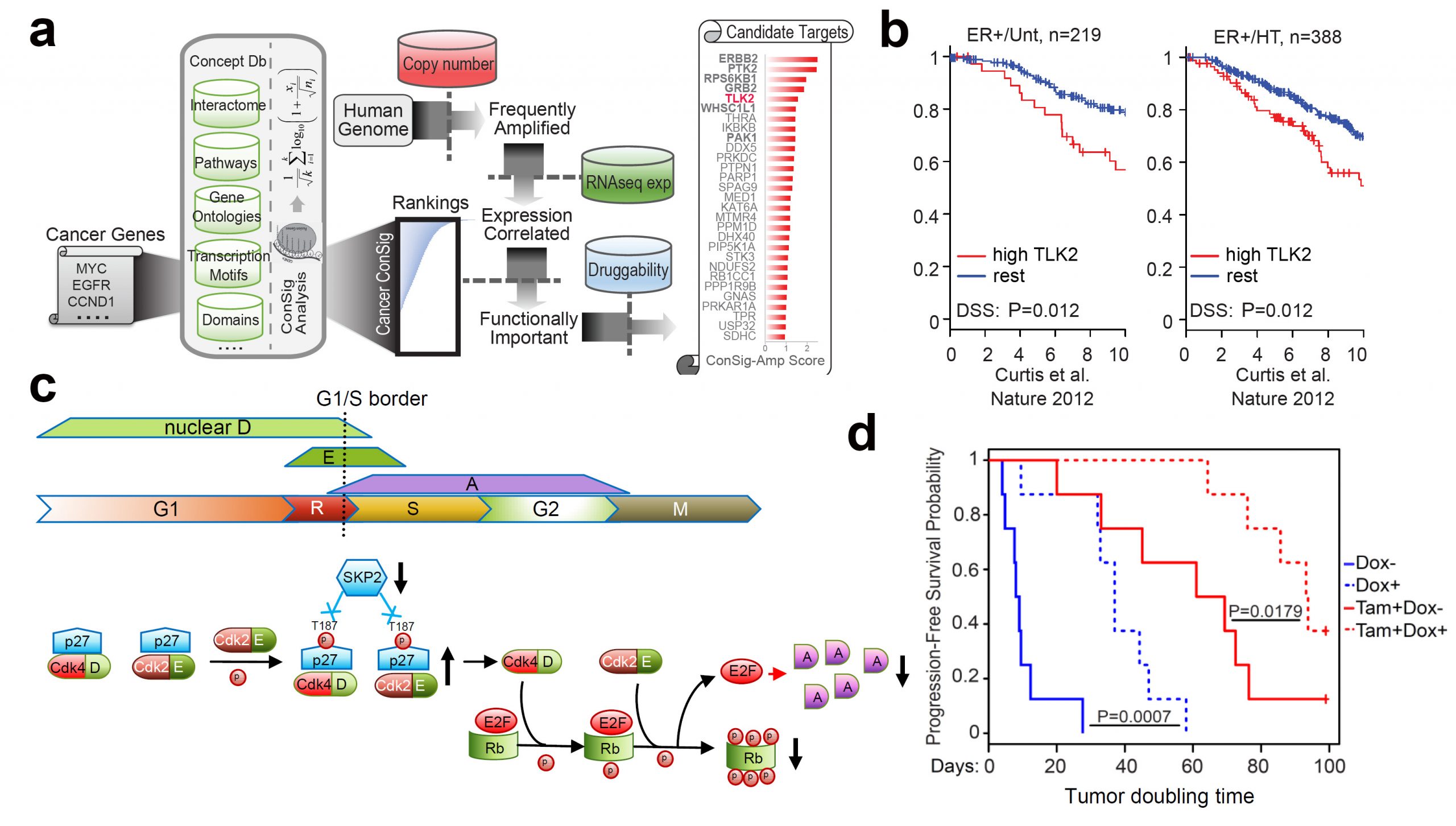
Identification of TLK2 as an amplified kinase target in aggressive luminal breast cancer. (a) The bioinformatics workflow of ConSig-Amp to discover therapeutically relevant oncogene targets in cancer at genome-wide scale based on TCGA copy number and RNAseq datasets. (b) Kaplan-Meier plots based on multiple gene expression datasets showing correlation of TLK2 overexpression with the outcome of systemically untreated or endocrine-treated ER+ breast cancer patients. (c) A schematic of normal G1/S cell cycle signaling and their alternations following TLK2 inhibition (black arrows). (d) The effect of TLK2 inhibition in the MCF7 xenograft tumors inducibly expressing a TLK2 shRNA, in the presence or absence of concomitant tamoxifen treatment. Figure shows the Kaplan–Meier survival plot comparing the progression-free survival of different treatment groups.
B. Therapeutic targeting of nemo-like kinase in primary and acquired endocrine-resistant breast cancer
In addition to TLK2, we also identified Nemo-Like Kinase (NLK) as a new actionable kinase target that endows previously uncharacterized survival signaling in endocrine resistant breast cancer. Mechanistically, NLK may function at last in part via enhancing the phosphorylation of ERα and its key coactivator SRC-3 to modulate ERα transcriptional activity, leading to reduced endocrine responsiveness and worse outcome of tamoxifen-treated patients. Through interrogation of a kinase-profiling database, we uncovered and verified a highly selective dual p38/NLK inhibitor, VX-702. Co-administration of VX-702 with the mTOR inhibitor Everolimus demonstrated a significant therapeutic effect in cell line- and patient-derived xenograft tumor models of acquired or de novo endocrine resistance (Clinical Cancer Research 2021. Read More.).
C. Identification of novel diagnostic urine biomarkers for urothelial carcinoma and discovery of the UCA1 gene, one of the most studied long non-coding RNA in cancer.
Furthermore, we identified two diagnostic urine biomarkers UCA1 and UPK3A for sensitive and specific detection of bladder cancers. Among these, UCA1 is one of the most commonly studied long non-coding RNA (lncRNA) first cloned and named by Dr. Wang (Clinical Cancer Research. 2006. Read More.). This long non-coding RNA was later found to be a key oncogene in the tumorigenesis of multiple cancer types and has been widely studied worldwide (a search of google scholar resulted in 11600 publications).
Multi-omics based precision oncology
A. An integral genomic signature approach for tailored cancer therapy using genome-wide sequencing data
With the advent of low-cost sequencing, transcriptome and genome sequencing is expected to become clinical routine and transform precision oncology within the next decade. But there is a paucity of viable genome-wide modeling methods that can facilitate rational selection of patients for tailored intervention. Machine learning methods are predominantly developed from image and language processing, which lack specialized algorithms in design to deal with the hallmark characteristics of genomic data. Here we propose that the redundancies within high-dimensional features may help overcome sequencing errors and bias, a concept like the use of redundant steel rods to reinforce the pillars of a building. We define an integral genomic signature (iGenSig) as an integral set of redundant high-dimensional genomic features predicting therapeutic response. iGenSig modeling generates prediction scores based on the set of redundant genomic features from labeled genomic datasets of therapeutic responses, and then reduce the effect of feature redundancy via adaptively penalizing the redundant features detected in specific samples based on their co-occurrence computed from unlabeled genomic datasets for large tumor cohorts. With this approach, if a subset of genomic features was lost due to sequencing biases or experimental variations, redundant genomic features will sustain the prediction. Whereas the unbiased genomic information acquired from unlabeled large cancer cohorts will substantially improve cross-dataset applicability of the models. iGenSig modeling diminishes false positives resulting from sequencing errors and overweighing via averaging the weights of genomic features from specific samples, and prevents overfitting via dynamically adjusting the feature weights for training subjects (Nature Communications 2022. Read More.).
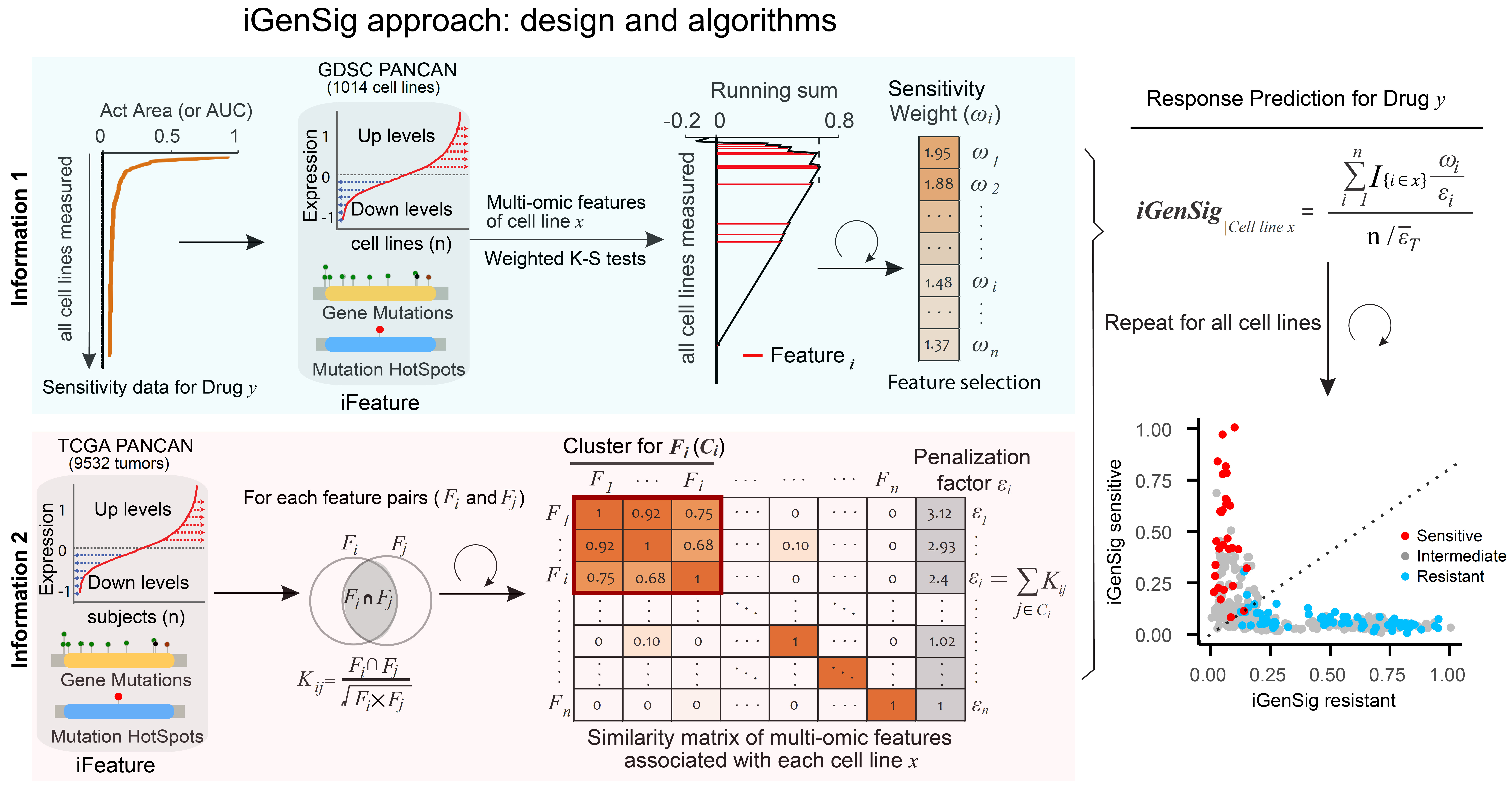
The workflow and algorithms of integral genomic signature analysis. The upper panel shows the calculation of the weights for significant genomic features that predict drug sensitivity or resistance based on weighted K-S tests of Act Area or AUC for each drug respectively, and the lower panel shows the computation of a similarity matrix for genomic features based on TCGA Pan-Cancer dataset to penalize the redundancy between the genomic features associated with each cell line x. The resulting sensitive or resistant genomic signature scores are calculated using the indicated formula based on the K-S tests using Act Area or AUC respectively. The dot plot shows the sensitive and resistant iGenSig scores for all cell line subjects.
Using genomic dataset of chemical perturbations, we develop a battery of iGenSig models for predicting cancer drug responses, and validate the models using independent cell-line and clinical datasets. The iGenSig models for five drugs demonstrate predictive values in six clinical studies. Via simple but best-fit algorithm design, iGenSig outperformed complex black-box machine learning methods in modeling therapeutic response on all six clinical trial datasets. The source code for iGenSig modeling is available through: https://github.com/wangxlab/iGenSig.
In the next, we further developed an integral genomic signature-based machine learning method called iGenSig-Rx as a white-box tool for modeling therapeutic response based on clinical trial datasets (https://github.com/wangxlab/iGenSig-Rx). We expect that iGenSig-Rx as a class of biologically interpretable multi-omics modeling methods will have broad applications in big-data based precision oncology. Read More.
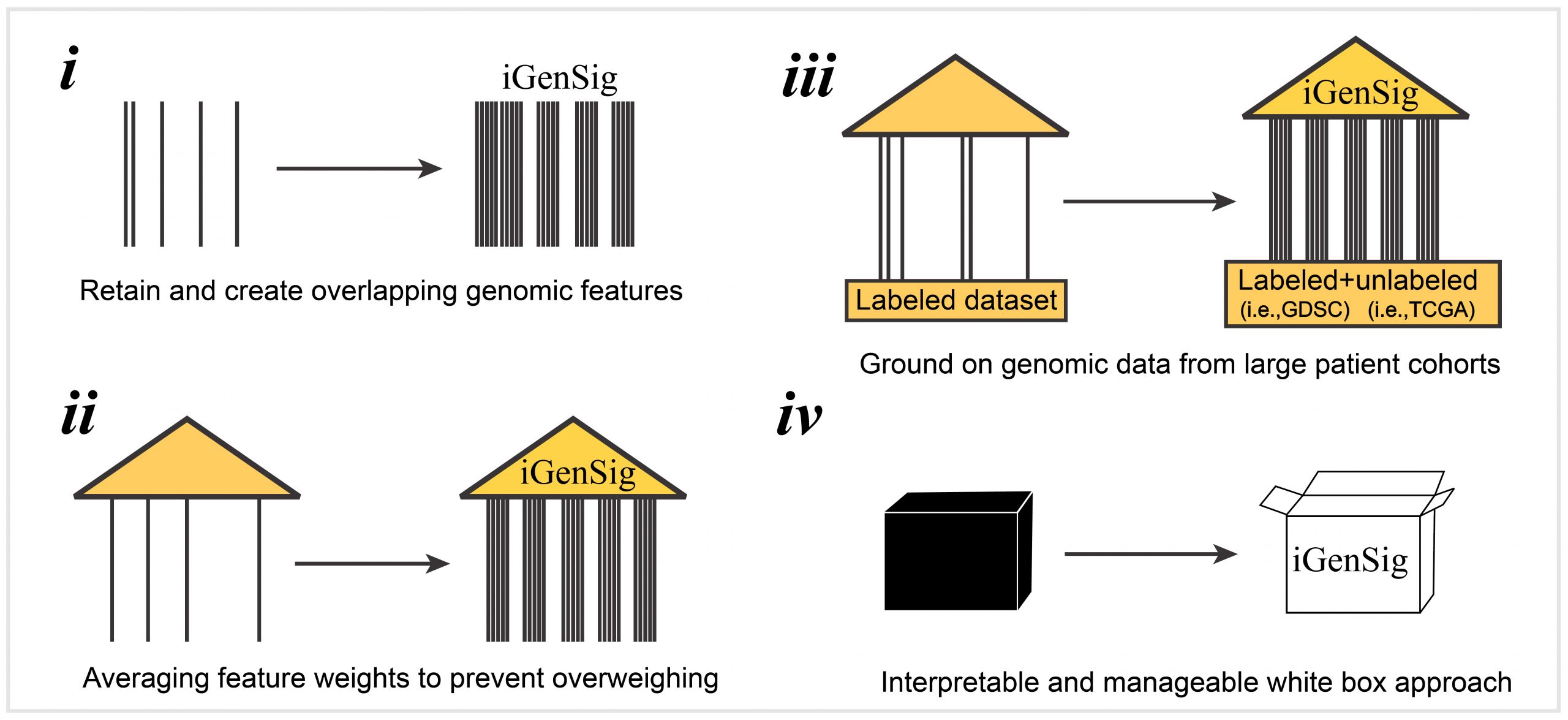
Schematic showing the principle and key features of iGenSig modeling: i) the iGenSig approach intentionally retains and creates redundant genomic features, a concept like the use of redundant steel rods to reinforce the pillars of a building. ii) iGenSig modeling utilizes the average correlation intensities of significant genomic features detected in specific samples to diminish the effect of false positive detection resulting from sequencing errors and prevent overweighing. iii) iGenSig modeling extract the second genomic information from unlabeled genomic datasets for large cohorts of human cancers, in addition to the labeled genomic datasets of drug sensitivity, which will substantially improve its cross-dataset applicability, particularly on clinical trial datasets. iv) iGenSig modeling is a white box approach, thus will be more interpretable and controllable than machine learning or deep learning approaches.
Furthermore, we have developed a R package called IndepthPathway — an integrated tool for in-depth pathway enrichment analysis based on bulk and single cell sequencing data (Read More). Leveraging its unique strength in deep functional interpretation, IndepthPathway will greatly enhance the application of bulk and single cell sequencing to explore the cellular pathway mechanisms at more precise resolution (https://github.com/wangxlab/indepthPathway).
Computational Immunogenomics for Precision immuno-oncology
A. Intragenic-rearrangement burden predicts immunotherapy response in tumor types with low TMB.
While immune checkpoint blockade (ICB) induces durable cancer remission, only a small subset of patients receive benefits, whereas a significant portion of patients encounter severe immune-related adverse events or hyperprogressive disease. While tumor mutation burden (TMB) differentiates responders in some cases, it is a weak predictor in tumor types with low mutation rates. Thus, there is an unmet need to discover a new class of genetic aberrations that predict ICB responses in these tumor types. Analyses of Pan-cancer whole genomes revealed that intragenic rearrangement (IGR) burden is a pivotal contributor to immune infiltration in breast, ovarian, esophageal, and endometrial cancers, which correlates with increased Macrophage M1 and CD8+ T cell signatures. Multivariate regression against spatially counted tumor infiltrated lymphocytes in breast, endometrial, and ovarian cancers suggest that IGR burden is a more influential covariate than other genetic aberrations in these cancers. In the MEDI4736 trial evaluating durvalumab in esophageal adenocarcinoma, IGR burden correlates with patient benefits. In the IMVigor210 trial evaluating atezolizumab in urothelial carcinoma, IGR burden increases with platinum exposure and predicts patient benefits for TMB-low, platinum-exposed tumors. Altogether, we demonstrate that IGR burden contributes to T-cell inflammation and predicts ICB benefits in TMB-low, IGR-dominant tumors, or in platinum-exposed tumors (Cancer Immunology Research 2023, In Press).
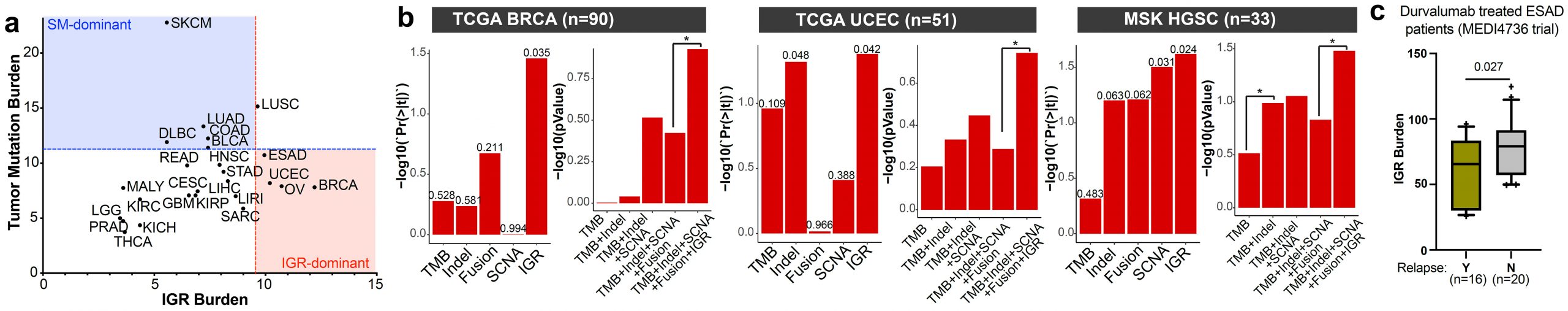
IGR burden is a pivotal contributor to T-cell inflammation in IGR dominant cancer types such as breast, uterine, ovarian, and esophageal cancers. (a) The landscape of IGR burden across all ICGC cancer types. (b) The correlations of genetic markers with spatial TIL counts in TCGA breast cancer (BRCA) (left), TCGA uterine corpus endometrial carcinoma (UCEC) (middle), or high-grade serous carcinomas (HGSC) of the MSK dataset (right). Tumors with matched WGS and spatial TIL count data are shown in the figure. Left panels, the p-value for each genetic marker in the multivariate model containing all genetic markers. Right panels, comparisons of the composite models containing different genetic markers. *p<0.05. (c) Boxplot comparing IGR burdens in the ESAD patients with or without cancer relapse in the MEDI4736 trial testing durvalumab.
This finding explains the following clinical aspects: In triple-negative breast cancer, high tumor mutation burden (TMB) only accounts for 5-10% of patients, whereas a much larger proportion of tumors are inflamed (26-50%), suggesting an uncharacterized major source of neoantigens. Similarly, TMB is not predictive of ICB response in ovarian cancers according to the IMagyn050 trial. In esophageal cancer, TMB is not associated with T cell infiltration and does not predict ICB response. This suggests that IGR burden may be a major source of neoantigens in these cancer types and supports the importance to include this new genetic marker in the sequencing panel for these cancers. Thus, we expect this study could have a similar clinical impact in IGR-dominant cancers as the study of tumor mutation burden in simple mutation-dominant cancers.
B. Identification of tumor-associated antigen targets
Tumor specific antigens (TSAs) have been widely adopted in clinics as active diagnostic and therapeutic targets in cancer. In our previous research project aimed at genome-wide detection of immunological targets, we analyzed the antigens widely adopted as clinical targets, and observed that these antigens usually present a distinctive heterogeneous gene expression profile in large-scale microarray datasets (Figure 5a). We therefore developed the Heterogeneous Expression Profile Analysis (HEPA) which preferentially identifies the clinically useful tumor antigens from the human genome. We then evaluated the immunogenicity of the TSAs by detecting specific autoantibodies in cancer patients. To deal with the large number of candidates, we developed a novel assay called Protein A/G based Reverse Serological Evaluation (PARSE), in which radio-labeled, in vitro translated proteins were used as probes for the presence of serum antibodies (Figure 5b). This allows for quick detection of the autoantibodies against a wide array of serum samples without the need of producing purified recombinant proteins. Further, in this assay, the in vitro translated tumor antigens retained the natural protein conformation and post- translational modifications, thus generating a precise picture of autoantibody responses against these antigens in cancer. Seven out of twelve novel antigens evaluated by PARSE elicited highly tumor-specific autoantibody responses in 4-15% of patients with selected cancers, resulting in distinctive autoantibody signatures in lung and stomach cancers.
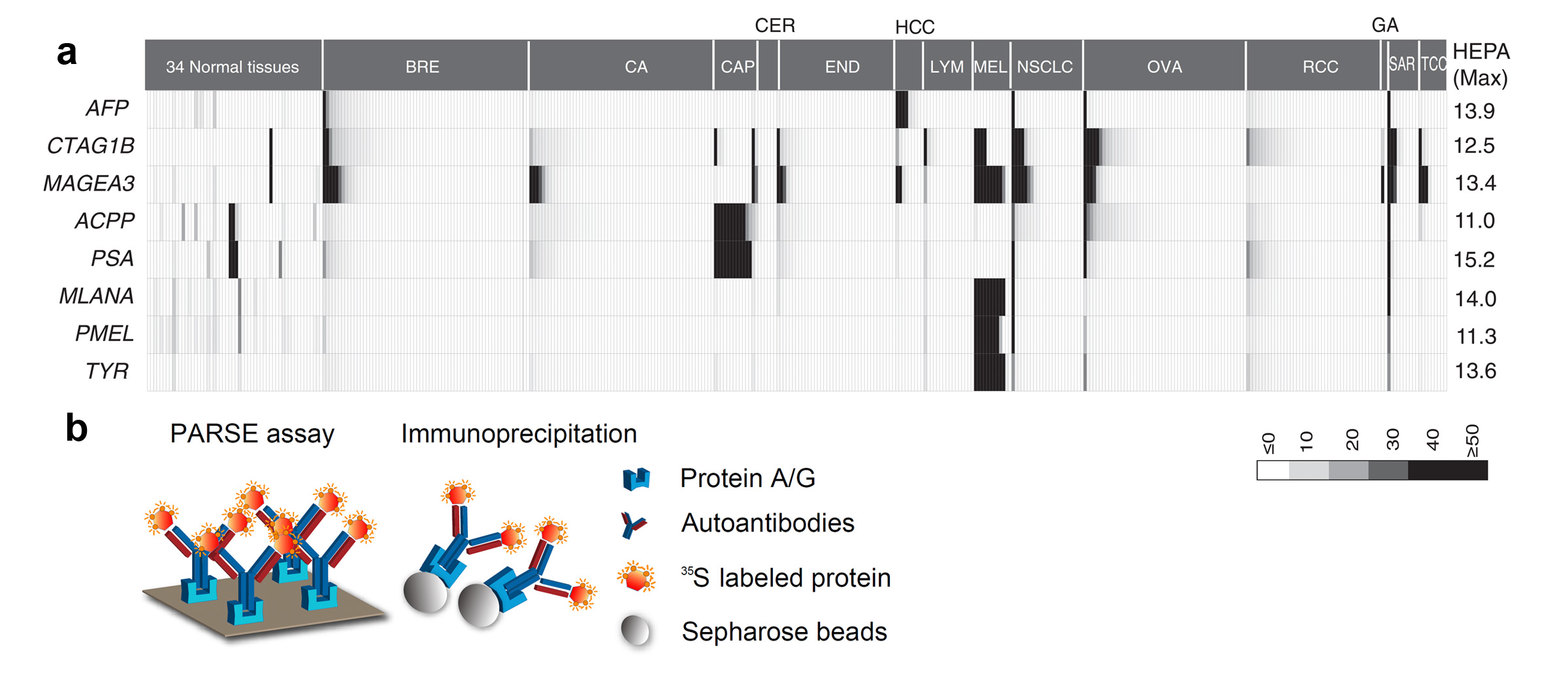
HEPA and PARSE: Systematic discovery of
clinically relevant tumor-specific antigens (TSA). (a) The heterogeneous expression profile of clinically
important tumor-specific antigens that provided the foundation for
heterogeneous expression profile analysis (HEPA). (b) Protein A/G–based reverse
serological evaluation (PARSE) for the quick detection of serum autoantibodies
against an array of putative TSA genes.